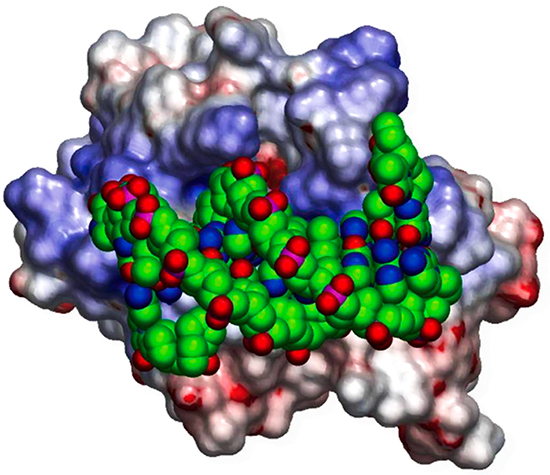
Our research starts from the hypothesis that sequence and structural information combined with biophysical analysis and machine learning can reveal the fundamental physical principles that underlie a wide range of biological phenomena. We currently have research programs focusing on: a) the integration of structural biology and systems biology through the prediction of protein-protein interactions; b) how highly specific protein-protein interactions mediate the wiring of neural circuits; c) optimizing antibodies using free energy perturbation methods. Our work includes theoretical research, biophysical measurements, the development of software tools, and specific applications to problems of biological importance. In the past we have elucidated the structural and energetic origins of protein-protein, protein-nucleic acid, and protein-membrane interactions, developed methods for protein structure prediction, and detected novel structural and functional relationships between proteins based on their geometric similarity. Our earlier work on the electrostatic properties of proteins has yielded widely used software tools and graphical depictions of protein surfaces that have become staples of the structural and computational biology communities.
Barry HonigBarry Honig
Laboratory of Molecular Biophysics




